Volume: 19 Issue: 9 September 2021
FULL TEXT
Abstract
We describe a case of a 55-year-old woman with polycystic kidney disease who received a living donor kidney transplant 16 years earlier and was on immunosuppressive therapy with satisfactory renal function. The donor was her mother. The patient presented with flank pain on the right side and macrohematuria, and noncontrast computed tomography and magnetic resonance imaging led to the diagnosis of tumors in the remaining right native polycystic kidney and ureter, as well as secondary retroperitoneal dissemination. We performed right radical nephrectomy and ureterectomy with extirpation of 2 metastases; the left native kidney remained intact. Histology showed squamous metaplastic changes and invasive epithelial neoplasm in the lumen of the renal pelvis and ureter with extensive squamous differentiation positive for nuclear p63 as squamous cell immunohistochemical marker. After surgery, an immunosuppressive therapy with methylprednisolone was administered, without calcineurin inhibitors and mycophenolate mofetil. Twelve months later the patient was still alive, with a glomerular filtration rate of 29 mL/min. Needs remain for further treatment modalities in patients with primary squamous cell carcinoma in nonfunctioning kidneys and improvements in imaging technique accuracy.
Key words : Imaging technique, Native kidney, Polycystic kidney disease
Introduction
Primary renal squamous cell carcinoma (SCC) is a rare event. It constitutes 0.5% to 0.8% of all renal neoplasms.1-3 Squamous cell carcinomas are usually diagnosed only in advanced tumor stage and associated with some anomalies such as staghorn calculi, ectopic kidneys, horseshoe kidneys, renal pelvis duplication, and polycystic kidney disease.4,5 The usual clinical presentation for SSC has no specific clinical features except the presence of urinary obstruction in all cases, chronic inflammation, and longstanding urothelial irritation. Squamous cell carcinomas are among the most aggressive malignant tumors, with early locoregional spread, metastasis, and poor prognosis. The majority of renal SCCs are nonfunctioning. The use of computed tomography (CT) and magnetic resonance imaging (MRI) with contrast media is generally contraindicated because of contrast media toxicity. Therefore, the precise diagnosis of SCC by standard imaging methods is often missed.6 Radical surgery is the most appropriate treatment. Adjuvant chemotherapy and radiotherapy are usually ineffective.
We present a very rare case of primary SCC in a patient with polycystic kidney disease, which occurred 16 years after living donor transplant. The donor was her mother. The SCC reported here is a unique case affecting native kidney and ureter concomitantly. To the best of our knowledge, such a case has not been reported previously.
Case Report
A 55-year-old female kidney transplant recipient was admitted at our hospital with macrohematuria and right lumbar flank pain. She was prescribed standard immunosuppressive therapy including cyclosporin, methylprednisolone, and mycophenolate mofetil. In the recent past years, the patient developed arterial hypertension, which was treated with calcium channel blockers. Thereafter, she was diagnosed with nephrotic syndrome and diabetes mellitus and treated with insulin and angiotensin-receptor blockers, with a partially beneficial effect on proteinuria.
Laboratory findings at admission were hemoglobin 7.0 g/dL, red blood cells 2.7 × 1012, hematocrit 23%, platelets 138 × 1012, estimated glomerular filtration rate 39.43 mL/min, and blood urea 20 mmol/L. Antibody tests were positive for hepatitis C virus and negative for hepatitis B surface antigen and HIV. Standard imaging procedures including echotomography, CT scan, and MRI, which showed a large polycystic kidney on the right side (126 × 70 mm) with a hyperdense formation probably corresponding to a large tumor mass (Figure 1A). There was no other obvious source of hemorrhage, so we decided to remove the native kidney, ie, a large polycystic kidney together with its perirenal fat tissue and fascia (Figure 2A).
After dissection of the removed kidney, a large, encapsulated solid mass of gray-yellow color was found between the middle and upper part of the kidney. It measured 3 × 3 × 2.5 cm and presented irregular zones of hemorrhage and necrosis (Figure 2B). Cyst diameters were 1 to 3.5 cm. The neoplasm had grown in the form of big patches and had passed the wall of the renal pelvis and spread upon it. There were areas with mild to severe squamous dysplasia and zones of keratin pearls, suggestive of squamous malignant metaplasia of the renal pelvis mucosa on the surrounding kidney parenchyma with signs of lymphatic and vascular invasion (Figure 3A, 3B and 3C). The kidney fascia was intact, but renal blood vessels were filled with malignant cells. On immunohistochemistry examination, the neoplastic tissue stained positive for keratin marker p63 (Figure 3D). After the operation, the patient was dismissed from the hospital in a satisfactory clinical condition, without hematuria and with satisfactory renal function with estimated glomerular filtration rate of 36 mL/min.
Four months later, the patient returned to our clinic with macroscopic hematuria. Various investigations including MRI revealed the presence of a double wall and infiltration of a large distal portion of the right native ureter (Figure 1B). Cystoscopy showed a large coagulum in the bladder with intermittent passage of blood from the native ureter. Further investigation of the distal part of the ureter by retrograde ureterorenoscopy showed a flat tumor. Beyond the tumor, the ureteral lumen was obstructed by coagulated blood, and the ureteral wall was inflamed and contained numerous urothelial erosions. Cytology done on ureterorenoscopy washing material confirmed the presence of highly malignant cells.7 Magnetic resonance imaging revealed 2 secondary retroperitoneal masses on the right-side lateral abdominal wall near the right-side liver lobe (Figure 1C).
Although the patient had advanced-stage cancer with hemorrhage, we decided to perform radical native ureterectomy and extirpation of the secondary masses (Figure 3E and 3F). The complexity of the operation resulted in a slow but successful rehabilitation of the patient. Given the highly malignant nature of metaplastic SCC, we decided to withhold proper immunosuppression (except methylprednisolone, 16 mg/day) after both the first and second operation. Twelve months later, the patient exhibited an estimated glomerular filtration rate of 29 mL/min, without hematuria.
Discussion
The occurrence of malignant degeneration or de novo carcinoma in patients with polycystic kidney disease is well known in kidney transplant recipients.8-10 Despite some controversy, it is generally acknowledged that the polycystic kidney represents a precancerous state. More than 35% of patients with polycystic kidney disease develop malignant degeneration of their kidneys as part of a multiorgan disease with the characteristics of neoplasia.9,10 The small number of reports on malignant disease in kidney transplant recipients with polycystic kidney may be explained by the fact that about 20% to 30% of these patients undergo nephrectomy before transplant.11 Based our personal experience, we strongly suggest preemptive nephrectomy in all patients with polycystic kidney disease.
Primary SCC is a rare and very aggressive tumor of the native kidney and ureter and is usually observed only late after renal transplant.2,12 The underlying mechanism is urothelial metaplasia and/or dysplasia of the pyelic and ureteral mucosa, with eventual transformation to SCC. Neoplasia may be primary or may be secondary to the metastasis of another primary SCC such as lung or skin carcinomas or genital malignancies in women.13 A tumor in a nonfunctioning kidney is difficult to diagnose because of the disorganized renal architecture and the diagnostic failure of the usual contrast media methods, which are contraindicated for this case. Therefore, additional diagnostic modalities such as MRI, positron emission tomography, CT-guided biopsy of tumor mass, ureterorenoscopy, and/or urinary cytology are required. Clinical signs and symptoms are infrequent and nonspecific. In our case, the occurrence of macroscopic hematuria and lumbar pain prompted us to conduct a detailed investigation.
The most important factor favoring the development of renal cell carcinoma in kidney transplant recipients is a long-term regimen of immunosuppression. The administration of calcineurin inhibitors and mycophenolate mofetil certainly favored the development of malignancy in our case, together with chronic inflammation and urinary obstruction. The patient had received cyclosporin A treatment for more than 10 years, with addition of tacrolimus for the last 6 years. Cyclosporin A is thought to favor carcinogenesis to a higher degree than tacrolimus.14 In our case, we first performed radical ablation of the native kidney, followed by a second step of ureterectomy with elimination of 2 major secondary retroperitoneal masses. Hematuria ceased immediately and the patient’s condition began to slowly improve. We found local invasion from the renal pelvis to the renal veins and arteries in the destroyed renal parenchyma but not in the perirenal space and fat tissue. However, the presence of 2 metastases showed that the initial process was very aggressive. The presence of ascites with a mucinous mass in the recto-uterine Douglas space containing high-grade malignant cells confirmed the advanced stage of the disease. Surgical removal is the only recommended treatment modality in such cases.12
Because of the patient’s serious clinical condition, which was characterized by the presence of anemia, diabetes mellitus, nephrotic syndrome, and graft damage, we decided against any chemotherapy or radiotherapy, as these treatment modalities do not lead to a significant survival benefit. Therefore, we decided to modify only the immunosuppressive treatment regimen.14
Conclusions
Our report documents the occurrence of SCC in the native kidney and ureter of a kidney transplant recipient with polycystic kidney disease on a long-term regimen of immunosuppression. Because the natural course of SCC is severe, we suggest frequent diagnostic and imaging testing in the follow-up of all patients with intact native polycystic kidneys and with congenital anomalies and/or urinary obstruction, chronic inflammation, horseshoe kidney, or duplicity of the pyelocaliceal system. Follow-up should include urinary cytology and urinary bladder tumor antigen assessment and, if necessary, high-resolution ultrasonography of the native kidneys once or twice a year with subsequent CT or MRI. The treatment of choice is radical nephroureterectomy. In kidney transplant recipients with advanced-stage SCC, only minimal immunosuppression should be used, without chemotherapy or radiotherapy, which may potentially allow better patient survival.
References:
- Mathur S, Rana P, Singh S, Goyal V, Sangwan M. Incidentally detected squamous cell carcinoma in non-functioning kidney presenting as multi-cystic mass. J Surg Case Rep. 2011;2011(9):8. doi:10.1093/jscr/2011.9.8
CrossRef - PubMed - Salehipour M, Dastgheib N, Hosseinzadeh M, et al. Primary renal pelvis and ureter squamous cell carcinoma (SCC): a rare case report and review of literature. Int Med Case Rep J. 2019;12:189-192. doi:10.2147/IMCRJ.S203283
CrossRef - PubMed - Jakes A, Jani P, Menon M, et al. Renal squamous cell carcinoma of a native kidney after renal transplant: a case report. Transplant Proc. 2016;48(1):259-261. doi:10.1016/j.transproceed.2015.12.015
CrossRef - PubMed - Singh V, Sinha RJ, Sankhwar SN, Mehrotra B, Ahmed N, Mehrotra S. Squamous cell carcinoma of the kidney, rarity redefined: case series with review of literature. J Cancer Sci Ther. 2010;2(4):55-56. doi:10.4172/1948-5956.1000028
CrossRef - PubMed - Zheng I, Alameddine M, Tan Y, et al. Collecting duct carcinoma of the native kidney in a renal transplant recipient. Case Rep Transplant. 2017;2017:4527104. doi:10.1155/2017/4527104
CrossRef - PubMed - El-Hachem G, Choueiry C, Al Hajj Obeid W, Chamseddine N. Renal squamous cell carcinoma: rare and aggressive variant of renal cancers. Cancer Bio Ther Oncol. 2017;1(2):6.
CrossRef - PubMed - Bindra R, Gupta S, Gupta N. Cytological diagnosis of squamous cell carcinoma of renal pelvis. J Cytol. 2010;27(2):76-77. doi:10.4103/0970-9371.70756
CrossRef - PubMed - Xie J, Zhang XB, Wang WZ, Li HZ. Case report of renal pelvis squamous cell carcinoma with tumor embolus in autosomal dominant polycystic kidney disease. Medicine (Baltimore). 2016;95(33):e4597. doi:10.1097/MD.0000000000004597
CrossRef - PubMed - Wetmore JB, Calvet JP, Yu AS, et al. Polycystic kidney disease and cancer after renal transplantation. J Am Soc Nephrol. 2014;25(10):2335-2341. doi:10.1681/ASN.2013101122
CrossRef - PubMed - Patel P, Horsfield C, Compton F, Taylor J, Koffman G, Olsburgh J. Native nephrectomy in transplant patients with autosomal dominant polycystic kidney disease. Ann R Coll Surg Engl. 2011;93(5):391-395. doi:10.1308/003588411X582690
CrossRef - PubMed - American Cancer Society. Cancer Facts and Figures 2014. Accessed September 8, 2015. http://www.cancer.org/research/cancerfactsstatistics/cancerfactsfigures
CrossRef - PubMed - Holmang S, Lele SM, Johansson SL. Squamous cell carcinoma of the renal pelvis and ureter: incidence, symptoms, treatment and outcome. J Urol. 2007;178(1):51-56. doi:10.1016/j.juro.2007.03.033
CrossRef - PubMed - Wang J, Wang L, Long L, Tao Q, Xu F, Luo F. Solitary renal metastasis from squamous cell carcinoma of the lung: a case report. Medicine (Baltimore). 2019;98(5):e14310. doi:10.1097/MD.0000000000014310
CrossRef - PubMed - Stallone G, Infante B, Grandaliano G. Management and prevention of post-transplant malignancies in kidney transplant recipients. Clin Kidney J. 2015;8(5):637-644. doi:10.1093/ckj/sfv054
CrossRef - PubMed

Volume : 19
Issue : 9
Pages : 981 - 985
DOI : 10.6002/ect.2021.0094
From the 1Zan Mitrev Clinical Hospital, the 2Medical Faculty, Saints Cyril and Methodius University, the 3University Clinic of Urology, and the 4Macedonian Academy of Sciences and Arts, Skopje, North Macedonia
Corresponding author: Zivko Popov, Bul. Krste Misirkov 2, 1000 Skopje, North Macedonia
E-mail: zivkopopov2000@yahoo.com
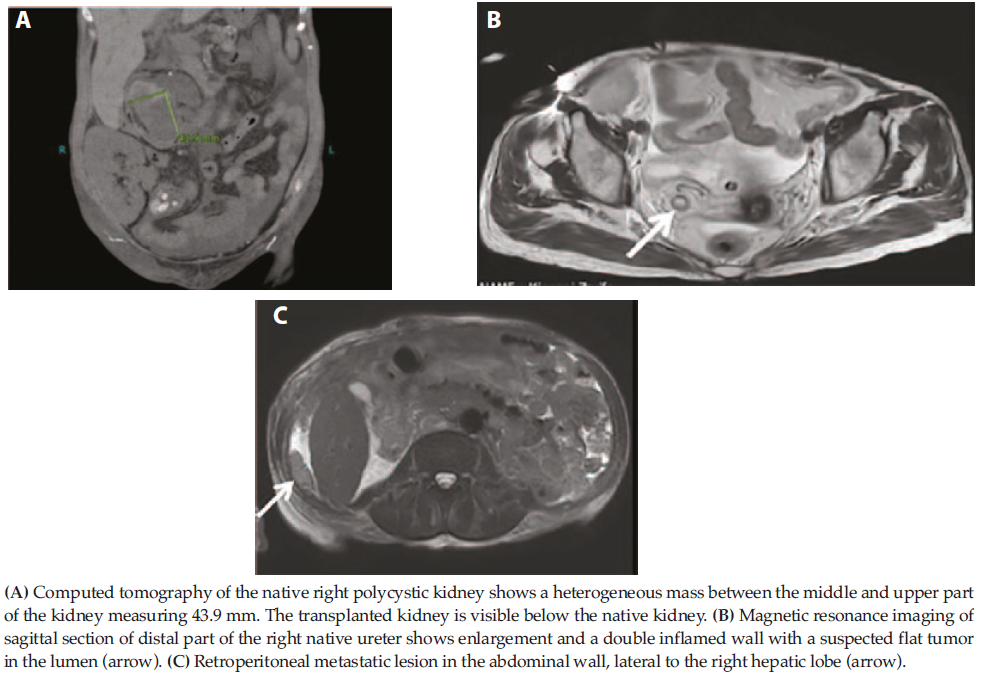
Figure 1. Computed Tomography and Magnetic Resonance Imaging of Native Polycystic Kidney, Ureter, and Abdominal Wall

Figure 2. Removal and Dissection of Native Polycystic Kidney With Perirenal Tissue and Fascia
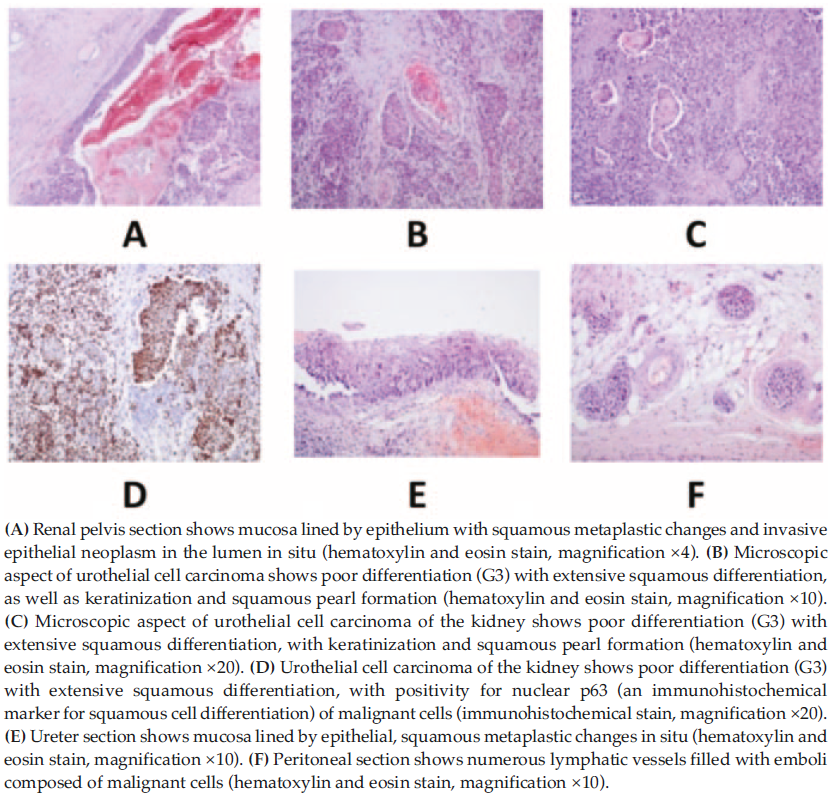
Figure 3. Renal Pelvis Section, Urothelial Cell Carcinoma, and Ureter and Peritoneal Sections